AtomSCF 教程总览
![]()
快速开始
本教程自动检测运行环境并安装依赖。执行下方第一个代码单元格即可开始!
学习目标
理解原子自洽场计算的完整逻辑链条:从 3D 原子问题到 1D 径向方程的求解。
完整计算流程
1. 物理模型简化
3D → 1D 约化:球对称势假设下,薛定谔方程分离变量,径向部分成为核心。
2. 数值离散化
连续 → 离散:径向网格系统(线性/对数)将微分方程转为矩阵本征问题。
3. 本征求解
求解器:五点有限差分法求解 Hamiltonian 矩阵,得到轨道波函数与能量。
4. 自洽迭代
SCF 循环:初始猜测 → 构造势 → 求解本征态 → 更新电子密度 → 检查收敛。
5. 方法分岔
HF vs DFT:
Hartree-Fock:显式交换积分(Slater 积分)
DFT-LDA:局域交换关联泛函(PZ81/VWN)
6. 自旋处理
LSDA vs LDA:自旋极化(LSDA)与自旋非极化(LDA)方法对比。
后续教程导航
01-grid.ipynb:径向网格系统(线性/对数网格)
02-solver.ipynb:本征值求解器(有限差分法)
03-scf.ipynb:自洽迭代流程
04-hartree-fock.ipynb:HF 交换算符与 Slater 积分
05-dft-xc.ipynb:DFT 交换关联泛函
06-spin.ipynb:自旋处理(LSDA/LDA)
07-complete-example.ipynb:端到端完整计算示例
理论基础:3D 到 1D 的约化
三维原子薛定谔方程在球对称势 \(V(r)\) 下可分离变量:
采用球坐标 \(\Psi(\mathbf{r}) = \frac{u(r)}{r}Y_{lm}(\theta, \phi)\),径向部分满足:
边界条件:\(u(0) = 0\),\(u(\infty) = 0\)(束缚态)
算法流程:网格离散 → Hamiltonian 矩阵构造 → 本征求解 → 波函数归一化
[1]:
# Colab 环境检测与安装
import sys
if "google.colab" in sys.modules:
!pip install -q git+https://github.com/bud-primordium/AtomSCF.git
[2]:
# 配置中文字体(避免乱码)
import matplotlib.pyplot as plt
import matplotlib
# 跨平台中文字体配置
matplotlib.rcParams['font.sans-serif'] = [
'Arial Unicode MS', # macOS
'WenQuanYi Micro Hei', # Linux
'SimHei', # Windows
'DejaVu Sans' # Fallback
]
matplotlib.rcParams['axes.unicode_minus'] = False
# 清除字体缓存(重要!)
try:
import matplotlib.font_manager as fm
fm._load_fontmanager(try_read_cache=False)
except Exception:
pass
快速示例:Al 原子 LSDA 计算
3 行代码展示完整流程:
[3]:
import numpy as np
from atomscf.grid import radial_grid_linear
from atomscf.scf import SCFConfig, run_lsda_pz81
# 1. 生成径向网格
r, w = radial_grid_linear(n=500, rmin=1e-5, rmax=30.0)
# 2. 配置 Al 原子 (Z=13,自动加载基态占据: 1s² 2s² 2p⁶ 3s² 3p¹)
cfg = SCFConfig(Z=13, r=r, w=w)
# 3. 运行 LSDA-PZ81 自洽计算
result = run_lsda_pz81(cfg)
# 4. 输出结果
print(f"收敛状态: {result.converged}")
print(f"迭代次数: {result.iterations}")
# 访问能量(注意检查 energies 是否为 None)
if result.energies:
E_total = result.energies.get("E_total", 0.0)
print(f"总能量: {E_total:.6f} Hartree")
print(f"总能量: {E_total * 27.211:.3f} eV")
print("\n轨道能量 (Hartree):")
# 正确的自旋索引:使用 "up" 和 "down" 字符串
for l in [0, 1]: # s, p
orbital_label = {0: "s", 1: "p", 2: "d"}[l]
# 自旋向上通道
energies_up = result.eps_by_l_sigma.get((l, "up"), [])
for n_idx, eps in enumerate(energies_up, start=1):
print(f" {n_idx}{orbital_label}(↑): {eps:.6f}")
# 自旋向下通道
energies_dn = result.eps_by_l_sigma.get((l, "down"), [])
for n_idx, eps in enumerate(energies_dn, start=1):
print(f" {n_idx}{orbital_label}(↓): {eps:.6f}")
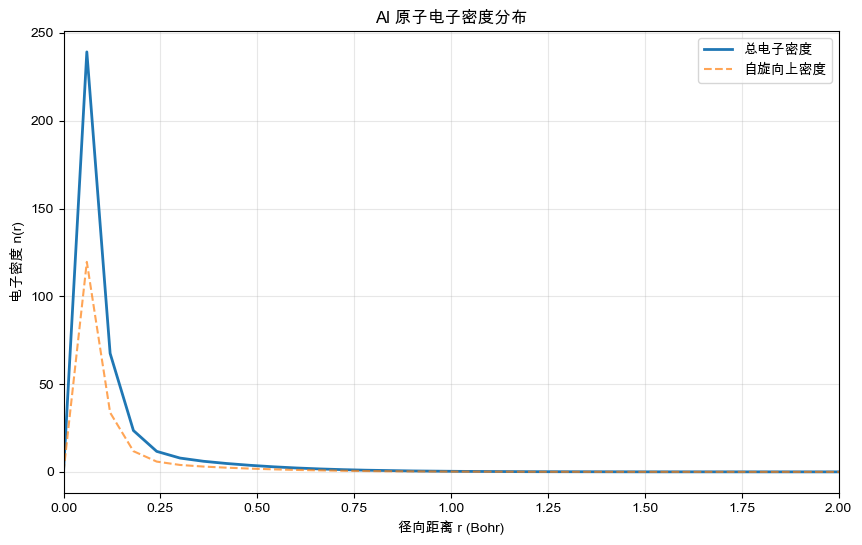
可视化:电子密度分布
[4]:
import matplotlib.pyplot as plt
plt.figure(figsize=(10, 6))
plt.plot(r, result.n_up + result.n_dn, label="总电子密度", linewidth=2)
plt.plot(r, result.n_up, "--", label="自旋向上密度", alpha=0.7)
# plt.plot(r, result.n_dn, "--", label="自旋向下密度", alpha=0.7)
plt.xlabel("径向距离 r (Bohr)")
plt.ylabel("电子密度 n(r)")
plt.title("Al 原子电子密度分布")
plt.xlim(0, 2)
plt.legend()
plt.grid(True, alpha=0.3)
plt.show()
核心概念预览
径向网格
网格点数与精度的平衡:500 点足够基准测试,2000 点用于高精度计算。
SCF 收敛
典型迭代次数:10-30 次(取决于初始猜测质量和混合参数)。
能量单位
AtomSCF 内部使用原子单位(Hartree),输出可转换为 eV:1 Hartree = 27.211 eV。
自旋索引
结果中的自旋通道使用字符串 "up" 和 "down" 索引,访问格式为 eps_by_l_sigma[(l, "up")]。
下一步
从 01-grid.ipynb 开始深入学习各模块!