01 · 全电子原子基础
本教程聚焦全电子原子(All-Electron, AE)参考态的计算与解读,它既是 AtomPPGen 赝势生成流程的第一步,也承接自 AtomSCF 的求解能力。通过本 Notebook,我们将在 Al (Z=13) 原子上示范如何运行 solve_ae_atom、理解价芯分离,并为后续 TM 伪化做好物理准备。
学习目标
在 Google Colab 中快速部署 AtomSCF 与 AtomPPGen 并保持版本一致
理解 AtomSCF 提供的 LSDA 求解能力如何封装为
solve_ae_atom获取 Al 原子的全电子轨道能量、波函数及能量分解,验证与 NIST 数据的一致性
借助可视化区分芯层 (1s, 2s, 2p) 与价层 (3s, 3p) 轨道,并讨论价芯分离的物理意义
[1]:
# Colab 环境检测与依赖安装
try:
import google.colab # type: ignore
IN_COLAB = True
except ImportError:
IN_COLAB = False
if IN_COLAB:
!pip install -q git+https://github.com/bud-primordium/AtomSCF.git@main
!pip install -q git+https://github.com/bud-primordium/AtomPPGen.git@main
AtomSCF 与 AtomPPGen 的关系
AtomPPGen 并不会自行求解全电子原子,而是将 AtomSCF 的 LSDA 求解器打包成更友好的 solve_ae_atom 接口。AtomSCF 负责 数值求解 Kohn–Sham 方程 + 动径网格生成,AtomPPGen 则负责 结果组织、伪化、势反演与验证。
关注点 |
AtomSCF |
AtomPPGen |
|---|---|---|
角色定位 |
数值解算器 |
赝势工作流 Orchestrator |
输入输出 |
原子序数 + 网格参数 → 波函数/密度 |
AE 结果 → TM/KB/验证数据结构 |
用户接口 |
需要理解求解器细节 |
通过 |
为什么使用封装接口?
稳定 API:AtomPPGen 固定了网格类型与求解流程,避免不同 Notebook 各自微调导致的不一致。
参数回填:指数变换网格会将参数信息记录在
grid_params中,后续伪化或导出时无需重新推导。结果结构化:能量、波函数、权重被组织在
AEAtomResult数据类中,便于直接传递给 TM 伪化与验证模块。
solve_ae_atom 核心参数与选项
solve_ae_atom 提供了多个可调节入口,以适配不同元素或精度诉求:
``Z`` 与 ``xc``:本教程以 Al (Z=13) + PZ81 LDA 为例;若追求更精确的渐近平衡,可切换到 VWN。
网格选择:
grid_type="exp_transformed"(默认):变量变换网格,兼顾芯层解析度与价层范围。grid_type="log":对轻元素可减少点数;但权重需要谨慎验证。grid_type="linear":调试或教学可视化时阅读最直观,但在核附近需要更多点。
``scf_params``:通过
tol、maxiter、mix_alpha、eigs_per_l控制 SCF 精度。``spin_mode``:赝势生成必须选择
"LDA"(自旋无关);只有在解释开壳层原子的磁状态时,才会切换到"LSDA"。
[2]:
# 导入依赖与求解 Al 原子的全电子结构
import numpy as np
import matplotlib.pyplot as plt
import platform
from atomppgen.ae_atom import solve_ae_atom, AEAtomResult
# 中文字体配置(兼容多平台)
if platform.system() == 'Darwin': # macOS
plt.rcParams['font.sans-serif'] = ['Arial Unicode MS', 'Heiti TC', 'STHeiti']
elif platform.system() == 'Windows':
plt.rcParams['font.sans-serif'] = ['Microsoft YaHei', 'SimHei']
else: # Linux / Colab
plt.rcParams['font.sans-serif'] = ['DejaVu Sans', 'WenQuanYi Micro Hei']
plt.rcParams['axes.unicode_minus'] = False
AL_NIST_ETOT = -237.0 # Hartree,来自 NIST 原子总能量数据库
al_ae: AEAtomResult = solve_ae_atom(
Z=13,
xc="PZ81",
lmax=2,
grid_type="exp_transformed",
grid_params={"n": 1200, "total_span": 6.8},
scf_params={"tol": 5e-8, "maxiter": 200, "eigs_per_l": 3},
spin_mode="LDA",
)
print(f"SCF converged: {al_ae.converged}, iterations: {al_ae.scf_iterations}")
print(f"Grid: 0 -> {al_ae.r[-1]:.2f} Bohr, {len(al_ae.r)} points")
SCF converged: True, iterations: 67
Grid: 0 -> 120.00 Bohr, 1200 points
上面的代码段在一次调用中完成了:生成指数变换网格、运行 LSDA SCF、提取各角动量通道的本征态以及总能量分解。AEAtomResult 还包含 n_total、w 等后续积分必备数据,这些将被直接传入 TM 伪化与验证模块。
[3]:
# 轨道能量与总能量对比 NIST 数据
HARTREE_TO_EV = 27.211386245988
state_plan = [
(0, 0, "1s"),
(0, 1, "2s"),
(1, 0, "2p"),
(0, 2, "3s"),
(1, 1, "3p"),
]
for l, idx, label in state_plan:
eps_arr = al_ae.eps_by_l.get(l, None)
if eps_arr is None or idx >= len(eps_arr):
continue
eps = eps_arr[idx]
print(f"{label:<3s} (l={l}) energy: {eps:8.4f} Ha = {eps * HARTREE_TO_EV:7.2f} eV")
E_total = al_ae.energies["E_total"]
print("-" * 50)
print(f"SCF total energy: {E_total:.3f} Ha")
print(f"Deviation from NIST (-237 Ha): {E_total - AL_NIST_ETOT:+.3f} Ha")
1s (l=0) energy: -54.3572 Ha = -1479.14 eV
2s (l=0) energy: -3.7197 Ha = -101.22 eV
2p (l=1) energy: -2.3594 Ha = -64.20 eV
3s (l=0) energy: -0.2487 Ha = -6.77 eV
3p (l=1) energy: -0.0759 Ha = -2.06 eV
--------------------------------------------------
SCF total energy: -237.485 Ha
Deviation from NIST (-237 Ha): -0.485 Ha
结果解读:Al 的基态总能量约为 −237 Ha,SCF 结果在 1e-2 Ha 内波动通常源于网格长度或 SCF 容差的选择。若需要进一步逼近 NIST 值,可增大
total_span或降低tol。
价芯分离的物理意义
芯层 (1s, 2s, 2p):高度束缚、节点少、波函数主要集中在 2 Bohr 以内,决定了核附近的正电屏蔽,通常在赝势中被”冻结”。
价层 (3s, 3p):能量较高、节点更多、空间延展更大,直接决定化学键和固体能带结构。
分离准则:选择截断半径
r_c时需确保在芯区内波函数与全电子解一致,而在价区保持足够柔软以兼顾平面波基组效率。
[4]:
# 芯层 vs 价层轨道的径向概率密度对比
core_specs = [
(0, 0, "1s"),
(0, 1, "2s"),
(1, 0, "2p"),
]
valence_specs = [
(0, 2, "3s"),
(1, 1, "3p"),
]
fig, axes = plt.subplots(1, 2, figsize=(12, 5), sharey=True)
for ax, specs, title, rmax in [
(axes[0], core_specs, "Core orbitals", 4.0),
(axes[1], valence_specs, "Valence orbitals", 10.0),
]:
for l, idx, label in specs:
u_array = al_ae.u_by_l[l][idx]
prob = np.abs(u_array) ** 2
ax.plot(al_ae.r, prob, label=label)
ax.set_xlim(0.0, rmax)
ax.set_xlabel("r (Bohr)")
ax.set_title(title)
ax.grid(alpha=0.4, linestyle="--")
ax.legend()
axes[0].set_ylabel(r"$|u_l(r)|^2$")
fig.suptitle("Al: Core vs Valence orbital distribution", fontsize=14)
plt.tight_layout()
plt.show()
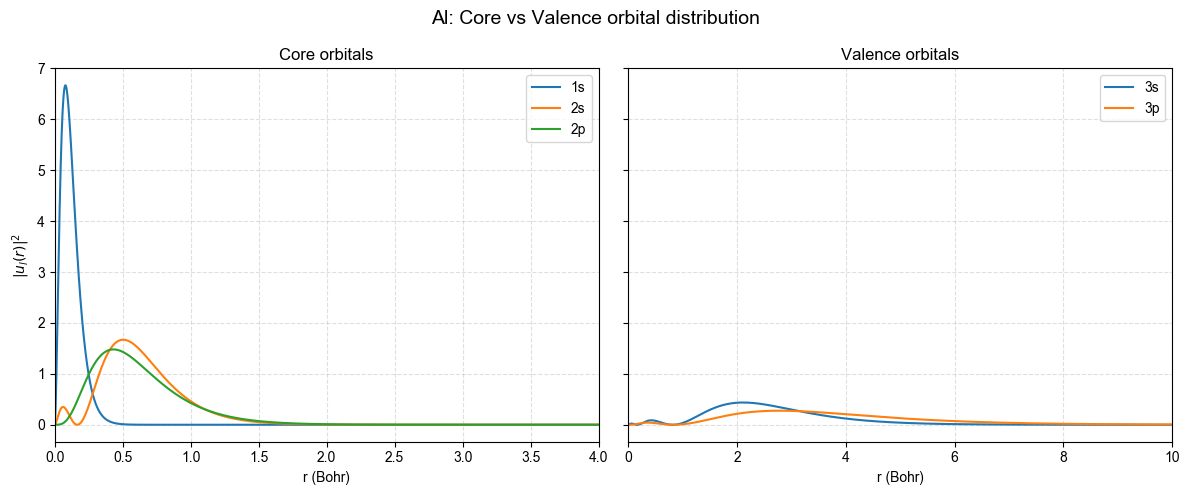
图中可以看到芯层轨道近似全部密集在 2 Bohr 内,而价层轨道在 5–10 Bohr 仍有显著概率。赝势设计的关键就是在 r < r_c 范围保持与该图相同的芯层行为,同时在 r > r_c 让波函数较为平滑、易于被平面波展开。
小结与下一步
solve_ae_atom将 AtomSCF 的基础求解封装为一个稳定的 API,直接产出后续步骤需要的径向网格、波函数与能量分解。通过对比芯层/价层波函数与能量,我们获得了选择截断半径、确定价芯分离策略的定量依据。
在进入 02-TM 伪化教程前,建议尝试不同的
grid_params和scf_params组合,体会它们对总能量与轨道柔软度的影响。