1. 弹性理论基础
![]()
1.1. 🚀 快速开始
本教程会自动检测运行环境:
Colab环境:自动安装ThermoElasticSim
本地环境:请确保已安装项目(参考README.md)
执行下方第一个代码单元格即可开始!
1.2. 学习目标
理解原子、晶胞和势函数的基本概念
掌握从原子力到连续介质应力的概念跳跃
学会通过静态计算获得弹性常数的基本原理
理解Hooke定律(Hooke's law)在原子尺度的体现
掌握应力-应变(stress-strain)关系和弹性张量的物理意义
1.3. 先备知识
基本的Python编程
线性代数基础(矩阵运算)
固体物理基础概念
1.4. 预计时长
执行时间:3-5分钟
学习时间:20-30分钟
1.5. 理论背景
本教程展示弹性理论的微观基础,详细的数学推导将在后续教程中介绍。
单位体系提示: 本教程使用 坐标(Å)、能量(eV)、应力(eV/ų→GPa) 单位体系
# Colab环境安装(仅在Colab中需要执行,Google免费硬件执行安装大约需要1分钟)
import sys
if "google.colab" in sys.modules:
!pip install -q git+https://github.com/bud-primordium/ThermoElasticSim.git
# 环境初始化(所有环境统一执行)
import numpy as np
import plotly.graph_objects as go
import plotly.io as pio
from scipy import stats
from thermoelasticsim.core.structure import Atom, Cell
from thermoelasticsim.potentials.eam import EAMAl1Potential
from thermoelasticsim.utils.plot_config import plt # 已设置Agg后端,避免Tk问题
from thermoelasticsim.utils.utils import EV_TO_GPA
from thermoelasticsim.visualization import api as viz
# 版本检查和基础设置
np.random.seed(42) # 确保结果可重现
try:
import thermoelasticsim
print(f"ThermoElasticSim版本: {thermoelasticsim.__version__}")
print(f"NumPy版本: {np.__version__}")
import scipy
print(f"SciPy版本: {scipy.__version__}")
except ImportError:
print("请确保已正确安装ThermoElasticSim")
raise
# 配置plotly为notebook交互显示
pio.renderers.default = "notebook_connected"
# matplotlib内联显示
%matplotlib inline
print("所有模块导入成功")
INFO: 字体配置成功,优先使用: Arial Unicode MS
ThermoElasticSim版本: 4.0.0
NumPy版本: 2.2.6
SciPy版本: 1.16.1
所有模块导入成功
1.6. 从单个原子到晶胞
我们从最基本的概念开始:什么是原子,什么是晶胞?
# 创建单个铝原子
al_atom = Atom(id=1, symbol="Al", mass_amu=26.98, position=[0, 0, 0])
print(f"原子信息:{al_atom.symbol}, 质量={al_atom.mass_amu:.2f} amu")
print(f"位置:{al_atom.position} Å")
print(f"速度:{al_atom.velocity} Å/fs")
print(f"受力:{al_atom.force} eV/Å")
# 创建简单的正交晶胞
lattice_constant = 4.05 # 铝的晶格常数 (Å)
lattice_vectors = np.eye(3) * lattice_constant
print(f"\n晶格矢量矩阵:\n{lattice_vectors}")
# 创建包含一个原子的晶胞
atoms = [al_atom]
simple_cell = Cell(lattice_vectors=lattice_vectors, atoms=atoms)
print(f"\n晶胞体积:{simple_cell.volume:.2f} ų")
print(f"原子数量:{simple_cell.num_atoms}")
原子信息:Al, 质量=26.98 amu
位置:[0. 0. 0.] Å
速度:[0. 0. 0.] Å/fs
受力:[0. 0. 0.] eV/Å
晶格矢量矩阵:
[[4.05 0. 0. ]
[0. 4.05 0. ]
[0. 0. 4.05]]
晶胞体积:66.43 ų
原子数量:1
★ 概念解释:
Atom:包含原子的所有信息(位置、速度、力等)Cell:定义了模拟盒子和周期性边界条件晶格矢量定义了盒子的形状和大小
# 3D可视化晶胞和原子
fig = viz.plot_structure_3d(simple_cell, show_box=True, title="单原子晶胞可视化")
fig.show()
print("提示:图形是交互式的,可以旋转、缩放查看")
提示:图形是交互式的,可以旋转、缩放查看
1.7. 构建小型FCC超胞
现在我们创建一个更真实的FCC(面心立方)结构,用于后续的势函数探索。
def create_fcc_supercell(lattice_constant, supercell_size):
"""创建FCC超胞"""
nx, ny, nz = supercell_size
# FCC基矢
lattice_vectors = np.array(
[
[lattice_constant * nx, 0, 0],
[0, lattice_constant * ny, 0],
[0, 0, lattice_constant * nz],
]
)
# FCC基原子位置(分数坐标)
fcc_basis = np.array(
[
[0.0, 0.0, 0.0], # 原点
[0.5, 0.5, 0.0], # xy面心
[0.5, 0.0, 0.5], # xz面心
[0.0, 0.5, 0.5], # yz面心
]
)
atoms = []
atom_id = 1
for i in range(nx):
for j in range(ny):
for k in range(nz):
for basis_pos in fcc_basis:
# 计算笛卡尔坐标
frac_pos = (basis_pos + [i, j, k]) / [nx, ny, nz]
cart_pos = frac_pos @ lattice_vectors
atom = Atom(
id=atom_id, symbol="Al", mass_amu=26.98, position=cart_pos
)
atoms.append(atom)
atom_id += 1
return Cell(lattice_vectors=lattice_vectors, atoms=atoms)
# 创建2×2×2的小超胞(32个原子)
small_cell = create_fcc_supercell(lattice_constant=4.05, supercell_size=(2, 2, 2))
print("小超胞信息:")
print(f"原子数量:{small_cell.num_atoms}")
print(f"体积:{small_cell.volume:.2f} ų")
print(f"密度:{small_cell.num_atoms / small_cell.volume:.4f} 原子/ų")
# 可视化小超胞
fig = viz.plot_structure_3d(small_cell, show_box=True, title="2×2×2 FCC铝超胞")
fig.show()
小超胞信息:
原子数量:32
体积:531.44 ų
密度:0.0602 原子/ų
1.8. 引入势函数概念
现在我们引入势函数的概念。势函数描述了原子间的相互作用,是分子动力学的核心。
# 创建EAM铝势
potential = EAMAl1Potential()
print(f"势函数类型:{potential.__class__.__name__}")
print(f"截断距离:{potential.cutoff} Å")
# 计算小超胞的初始能量
initial_energy = potential.calculate_energy(small_cell)
print(f"\n初始总能量:{initial_energy:.6f} eV")
print(f"每原子平均能量:{initial_energy / small_cell.num_atoms:.6f} eV/atom")
势函数类型:EAMAl1Potential
截断距离:6.5 Å
初始总能量:-112.565283 eV
每原子平均能量:-3.517665 eV/atom
def explore_potential_landscape():
"""探索势能随原子位移的变化"""
# 选择第一个原子进行微扰
test_atom_idx = 0
original_pos = small_cell.atoms[test_atom_idx].position.copy()
# 设置位移范围
displacements = np.linspace(-0.2, 0.2, 21) # -0.2到+0.2 Å
energies = []
print(f"扫描原子{test_atom_idx + 1}沿x方向的势能变化...")
for dx in displacements:
# 应用位移
small_cell.atoms[test_atom_idx].position[0] = original_pos[0] + dx
# 计算能量
energy = potential.calculate_energy(small_cell)
energies.append(energy)
# 恢复原位置
small_cell.atoms[test_atom_idx].position[0] = original_pos[0]
return displacements, energies
def explore_3d_potential_landscape():
"""探索x-y平面内的三维势能面"""
print("计算三维势能面...")
test_atom_idx = 0
original_pos = small_cell.atoms[test_atom_idx].position.copy()
# 创建二维网格
dx_range = np.linspace(-0.15, 0.15, 15) # x位移
dy_range = np.linspace(-0.15, 0.15, 15) # y位移
X, Y = np.meshgrid(dx_range, dy_range)
Z = np.zeros_like(X)
# 计算势能面
for i, dx in enumerate(dx_range):
for j, dy in enumerate(dy_range):
# 应用x-y位移
small_cell.atoms[test_atom_idx].position[0] = original_pos[0] + dx
small_cell.atoms[test_atom_idx].position[1] = original_pos[1] + dy
# 计算能量
energy = potential.calculate_energy(small_cell)
Z[j, i] = energy # 注意索引顺序
# 恢复原位置
small_cell.atoms[test_atom_idx].position[:2] = original_pos[:2]
return X, Y, Z
# 执行一维势能面扫描
displacements, energies = explore_potential_landscape()
# 绘制一维势能面
plt.figure(figsize=(10, 6))
plt.plot(displacements, energies, "bo-", markersize=6, linewidth=2)
plt.axhline(
y=min(energies),
color="red",
linestyle="--",
alpha=0.7,
label=f"最低能量: {min(energies):.6f} eV",
)
plt.axvline(x=0, color="green", linestyle="--", alpha=0.7, label="初始位置")
plt.xlabel("原子位移 (Å)", fontsize=12)
plt.ylabel("系统总能量 (eV)", fontsize=12)
plt.title("势能面概念:原子偏离初始位置的能量代价", fontsize=14)
plt.grid(alpha=0.3)
plt.legend()
plt.tight_layout()
plt.show()
# 分析一维结果
min_energy = min(energies)
min_idx = energies.index(min_energy)
energy_change = max(energies) - min_energy
print("\n一维势能面分析:")
print(f"最低能量位移:{displacements[min_idx]:.3f} Å")
print(f"能量变化范围:{energy_change:.6f} eV")
print(f"能量变化范围:{energy_change * 1000:.2f} meV")
# 执行三维势能面计算
X, Y, Z = explore_3d_potential_landscape()
# 使用plotly创建交互式三维势能面
fig = go.Figure(
data=[
go.Surface(
x=X, y=Y, z=Z, colorscale="Viridis", colorbar=dict(title="能量 (eV)")
)
]
)
fig.update_layout(
title="交互式三维势能面:原子在x-y平面的能量地形",
scene=dict(
xaxis_title="x位移 (Å)",
yaxis_title="y位移 (Å)",
zaxis_title="总能量 (eV)",
camera=dict(eye=dict(x=1.5, y=1.5, z=1.2)),
),
width=800,
height=600,
)
# 根据环境选择渲染方式
try:
if IN_COLAB:
fig.show()
else:
fig.show(renderer="notebook_connected")
except NameError:
fig.show()
# 创建等势线图作为2D补充
fig_contour = go.Figure(
data=go.Contour(
x=X[0, :],
y=Y[:, 0],
z=Z,
colorscale="Viridis",
contours=dict(showlabels=True, labelfont=dict(size=10, color="white")),
colorbar=dict(title="能量 (eV)"),
)
)
fig_contour.update_layout(
title="势能等高线图:能量在x-y平面的分布",
xaxis_title="x位移 (Å)",
yaxis_title="y位移 (Å)",
width=600,
height=500,
)
try:
if IN_COLAB:
fig_contour.show()
else:
fig_contour.show(renderer="notebook_connected")
except NameError:
fig_contour.show()
print("\n交互提示:")
print("• 三维图可以旋转、缩放来观察势能面的形状")
print("• 等高线图清楚显示了能量最小值的位置")
print("• 能量面的'山谷'对应稳定位置,'山峰'对应不稳定位置")
扫描原子1沿x方向的势能变化...
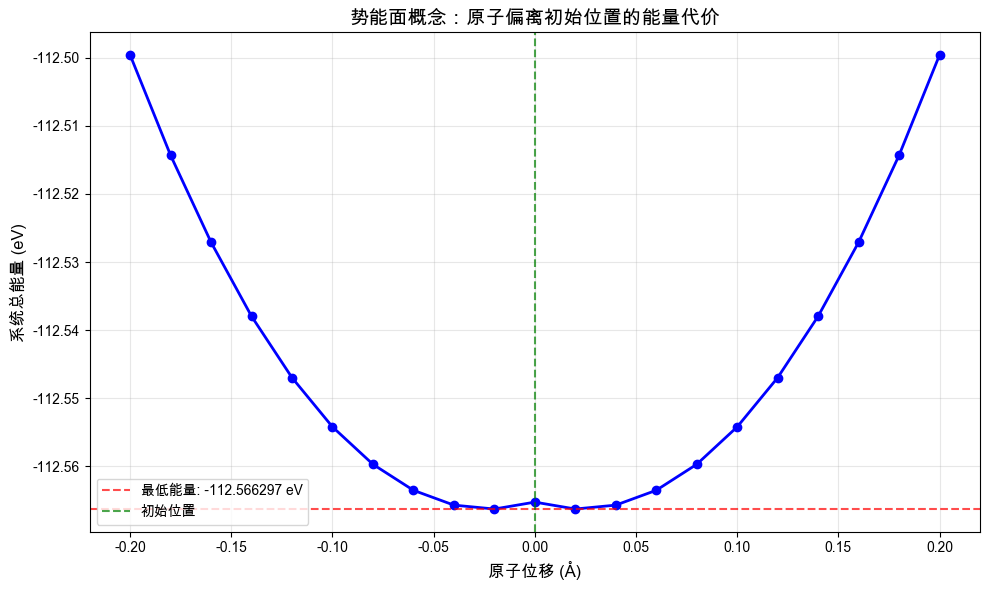
一维势能面分析:
最低能量位移:-0.020 Å
能量变化范围:0.066711 eV
能量变化范围:66.71 meV
计算三维势能面...
交互提示:
• 三维图可以旋转、缩放来观察势能面的形状
• 等高线图清楚显示了能量最小值的位置
• 能量面的'山谷'对应稳定位置,'山峰'对应不稳定位置
# 计算所有原子的受力
potential.calculate_forces(small_cell)
# 分析力的统计信息
forces = np.array([atom.force for atom in small_cell.atoms])
force_magnitudes = np.linalg.norm(forces, axis=1)
print("力统计信息(单位:eV/Å):")
print(f"最大力:{np.max(force_magnitudes):.6f}")
print(f"平均力:{np.mean(force_magnitudes):.6f}")
print(f"力的RMS:{np.sqrt(np.mean(force_magnitudes**2)):.6f}")
# 显示前5个原子的力
print("\n前5个原子的受力:")
for i in range(min(5, len(small_cell.atoms))):
force = small_cell.atoms[i].force
mag = np.linalg.norm(force)
print(
f"原子{i + 1}: [{force[0]:8.6f}, {force[1]:8.6f}, {force[2]:8.6f}] |F|={mag:.6f}"
)
# 如果力很小,说明结构接近平衡
if np.max(force_magnitudes) < 0.01:
print("\n✓ 结构接近平衡状态(最大力 < 0.01 eV/Å)")
else:
print(f"\n⚠️ 结构需要优化(最大力 = {np.max(force_magnitudes):.6f} eV/Å)")
力统计信息(单位:eV/Å):
最大力:0.159559
平均力:0.151585
力的RMS:0.151716
前5个原子的受力:
原子1: [-0.090046, -0.090046, -0.090046] |F|=0.155964
原子2: [-0.090046, -0.090046, -0.090046] |F|=0.155964
原子3: [-0.090046, -0.090046, -0.090046] |F|=0.155964
原子4: [-0.090046, -0.090046, -0.090046] |F|=0.155964
原子5: [-0.090046, -0.090046, 0.090046] |F|=0.155964
⚠️ 结构需要优化(最大力 = 0.159559 eV/Å)
1.9. 结构优化与零温基态搜索
通过优化晶格常数找到系统的零温基态。这是理解为什么需要结构优化的关键演示。
def optimize_lattice_constant():
"""优化晶格常数,绘制E-a曲线"""
# 测试不同的晶格常数
lattice_params = np.linspace(3.8, 4.3, 11)
energies = []
print("扫描不同晶格常数的能量...")
for a in lattice_params:
# 创建新的超胞
test_cell = create_fcc_supercell(lattice_constant=a, supercell_size=(2, 2, 2))
# 计算能量
energy = potential.calculate_energy(test_cell)
energy_per_atom = energy / test_cell.num_atoms
energies.append(energy_per_atom)
print(f"a = {a:.2f} Å → E = {energy_per_atom:.6f} eV/atom")
return lattice_params, energies
# 执行晶格优化
lattice_params, energies = optimize_lattice_constant()
# 找到最优值
min_idx = np.argmin(energies)
optimal_a = lattice_params[min_idx]
min_energy = energies[min_idx]
print("\n🎯 优化结果:")
print(f"最优晶格常数:{optimal_a:.2f} Å")
print(f"最低每原子能量:{min_energy:.6f} eV/atom")
print("实验值比较:Al的实验晶格常数 ≈ 4.05 Å")
扫描不同晶格常数的能量...
a = 3.80 Å → E = -3.363471 eV/atom
a = 3.85 Å → E = -3.428686 eV/atom
a = 3.90 Å → E = -3.474233 eV/atom
a = 3.95 Å → E = -3.502267 eV/atom
a = 4.00 Å → E = -3.516048 eV/atom
a = 4.05 Å → E = -3.517665 eV/atom
a = 4.10 Å → E = -3.508079 eV/atom
a = 4.15 Å → E = -3.487882 eV/atom
a = 4.20 Å → E = -3.457807 eV/atom
a = 4.25 Å → E = -3.418963 eV/atom
a = 4.30 Å → E = -3.372863 eV/atom
🎯 优化结果:
最优晶格常数:4.05 Å
最低每原子能量:-3.517665 eV/atom
实验值比较:Al的实验晶格常数 ≈ 4.05 Å
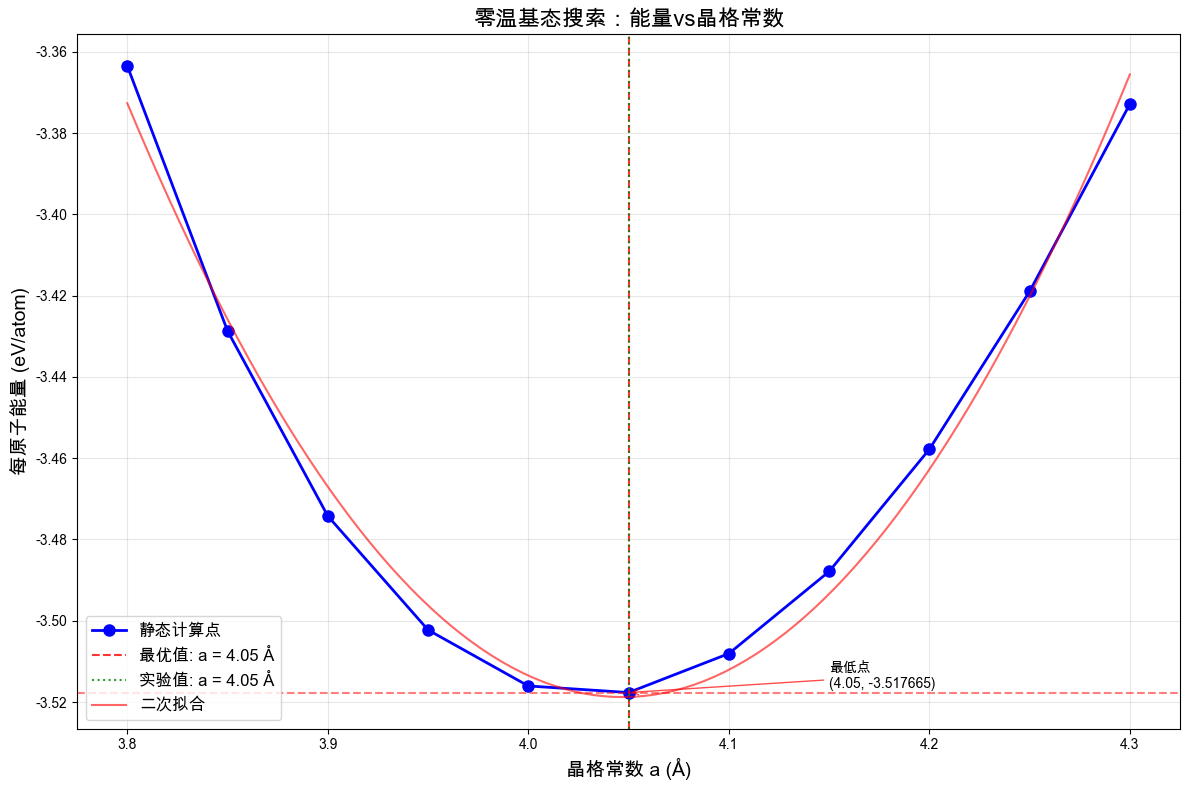
# 绘制能量-晶格常数关系图(这是理解零温基态概念的关键图)
plt.figure(figsize=(12, 8))
# 主图
plt.plot(lattice_params, energies, "bo-", markersize=8, linewidth=2, label="静态计算点")
# 标记最优点
plt.axvline(
optimal_a,
color="red",
linestyle="--",
alpha=0.8,
label=f"最优值: a = {optimal_a:.2f} Å",
)
plt.axhline(min_energy, color="red", linestyle="--", alpha=0.5)
# 标记实验值
plt.axvline(4.05, color="green", linestyle=":", alpha=0.8, label="实验值: a = 4.05 Å")
# 拟合抛物线
coeffs = np.polyfit(lattice_params, energies, 2)
poly = np.poly1d(coeffs)
a_fine = np.linspace(3.8, 4.3, 100)
plt.plot(a_fine, poly(a_fine), "r-", alpha=0.6, label="二次拟合")
plt.xlabel("晶格常数 a (Å)", fontsize=14)
plt.ylabel("每原子能量 (eV/atom)", fontsize=14)
plt.title("零温基态搜索:能量vs晶格常数", fontsize=16)
plt.grid(alpha=0.3)
plt.legend(fontsize=12)
# 添加注释
plt.annotate(
f"最低点\n({optimal_a:.2f}, {min_energy:.6f})",
xy=(optimal_a, min_energy),
xytext=(optimal_a + 0.1, min_energy + 0.001),
arrowprops=dict(arrowstyle="->", color="red", alpha=0.7),
fontsize=10,
ha="left",
)
plt.tight_layout()
plt.show()
# 计算体弹性模量的粗略估计
# K = V * d²E/dV² = (9/4) * a * d²E/da² (对于立方晶体)
second_derivative = 2 * coeffs[0] # 二次项系数的2倍
bulk_modulus_estimate = (9 / 4) * optimal_a * second_derivative
print(f"\n📐 体弹性模量粗略估计:{bulk_modulus_estimate:.1f} GPa")
print("实验值比较:Al的体弹性模量 ≈ 76 GPa")
print(f"误差:{abs(bulk_modulus_estimate - 76) / 76 * 100:.1f}%")
print("\n⚠️ 重要说明:")
print("• 本估计基于粗糙的11个采样点,精度有限")
print("• 小超胞(32原子)存在尺寸效应,影响精度")
print("• 没有进行内部原子弛豫,仅为刚性形变计算")
print("• 下一章将介绍精确的零温基态优化和原子弛豫方法")
print("• 如需高精度计算,请参考下节教程中的StructureRelaxer")
📐 体弹性模量粗略估计:43.6 GPa
实验值比较:Al的体弹性模量 ≈ 76 GPa
误差:42.6%
⚠️ 重要说明:
• 本估计基于粗糙的11个采样点,精度有限
• 小超胞(32原子)存在尺寸效应,影响精度
• 没有进行内部原子弛豫,仅为刚性形变计算
• 下一章将介绍精确的零温基态优化和原子弛豫方法
• 如需高精度计算,请参考下节教程中的StructureRelaxer
★ 深入理解:
能量-晶格常数曲线展示了零温基态的概念
最低点对应平衡晶格常数和基态能量
曲线的曲率与体弹性模量相关
这就是为什么需要结构优化的原因:找到能量最低的稳定构型
1.10. 从原子力到连续介质应力
现在我们进行关键的概念跳跃:从微观的原子力过渡到宏观的应力张量。
# 使用优化的晶格常数创建基态超胞
equilibrium_cell = create_fcc_supercell(
lattice_constant=optimal_a, supercell_size=(2, 2, 2)
)
# 计算基态的应力张量
potential.calculate_forces(equilibrium_cell)
stress_atomic = equilibrium_cell.calculate_stress_tensor(potential) # 单位:eV/ų
# 转换为GPa,使用标准常量
stress_gpa = stress_atomic * EV_TO_GPA
print("🔬 微观尺度:原子应力张量")
print("应力张量 (GPa):")
for i in range(3):
row = [f"{stress_gpa[i, j]:8.3f}" for j in range(3)]
print(f" [{' '.join(row)}]")
# 检查对称性和迹
print("\n📊 应力张量分析:")
print(
f"对角元(正应力):σ11={stress_gpa[0, 0]:.3f}, σ22={stress_gpa[1, 1]:.3f}, σ33={stress_gpa[2, 2]:.3f} GPa"
)
print(
f"非对角元(剪应力):σ12={stress_gpa[0, 1]:.3f}, σ13={stress_gpa[0, 2]:.3f}, σ23={stress_gpa[1, 2]:.3f} GPa"
)
print(f"迹(静水压力×3):tr(σ) = {np.trace(stress_gpa):.3f} GPa")
print(f"平均应力:σ_avg = {np.trace(stress_gpa) / 3:.3f} GPa")
if abs(np.trace(stress_gpa) / 3) < 0.1:
print("✓ 基态接近零应力状态")
else:
print(f"⚠️ 基态存在残余应力:{np.trace(stress_gpa) / 3:.3f} GPa")
🔬 微观尺度:原子应力张量
应力张量 (GPa):
[ 1.066 -0.154 -0.154]
[ -0.154 1.066 -0.154]
[ -0.154 -0.154 1.066]
📊 应力张量分析:
对角元(正应力):σ11=1.066, σ22=1.066, σ33=1.066 GPa
非对角元(剪应力):σ12=-0.154, σ13=-0.154, σ23=-0.154 GPa
迹(静水压力×3):tr(σ) = 3.197 GPa
平均应力:σ_avg = 1.066 GPa
⚠️ 基态存在残余应力:1.066 GPa
★ 概念跳跃:
原子尺度:每个原子受到周围原子的力
连续介质尺度:通过维里定理得到局部应力张量
应力张量:描述材料内部每点的应力状态
对角元:拉伸/压缩应力;非对角元:剪切应力
1.11. 应变的概念和可视化
应变描述材料的几何形变。我们通过施加微小形变来理解应变的概念。
def apply_uniaxial_strain_manual(cell, strain_amplitude):
"""手动实现单轴应变(教学演示用)"""
# 构造应变矩阵(单轴x方向)
strain_matrix = np.array([[strain_amplitude, 0, 0], [0, 0, 0], [0, 0, 0]])
# 应用形变:F = I + ε
deformation_gradient = np.eye(3) + strain_matrix
# 变形晶格矢量
new_lattice = cell.lattice_vectors @ deformation_gradient
# 变形原子位置(保持分数坐标不变)
fractional_coords = np.array(
[atom.position for atom in cell.atoms]
) @ np.linalg.inv(cell.lattice_vectors)
new_positions = fractional_coords @ new_lattice
# 创建新的形变晶胞
new_atoms = []
for i, atom in enumerate(cell.atoms):
new_atom = Atom(
id=atom.id,
symbol=atom.symbol,
mass_amu=atom.mass_amu,
position=new_positions[i],
)
new_atoms.append(new_atom)
return Cell(lattice_vectors=new_lattice, atoms=new_atoms)
def apply_uniaxial_strain(cell, strain_amplitude):
"""使用Cell.apply_deformation实现单轴应变"""
# 创建晶胞副本,避免修改原对象
import copy
strained_cell = copy.deepcopy(cell)
# 构造形变梯度矩阵:F = I + ε
deformation_gradient = np.eye(3)
deformation_gradient[0, 0] = 1 + strain_amplitude # x方向拉伸
# 使用Cell的标准方法应用形变(就地修改)
strained_cell.apply_deformation(deformation_gradient)
return strained_cell
# 应用不同程度的单轴应变
strain_values = [0.0, 0.003, 0.006] # 0%, 0.3%, 0.6%应变
print("应变演示:单轴拉伸")
# 确保使用已定义的基态晶胞
try:
base_cell = equilibrium_cell
print("使用优化的基态晶胞")
except NameError:
base_cell = create_fcc_supercell(
lattice_constant=optimal_a, supercell_size=(2, 2, 2)
)
equilibrium_cell = base_cell # 定义变量供后续使用
print("创建基态晶胞")
for strain in strain_values:
# 方法1:手动实现
strained_cell_manual = apply_uniaxial_strain_manual(base_cell, strain)
potential.calculate_forces(strained_cell_manual)
stress_manual = strained_cell_manual.calculate_stress_tensor(potential) * EV_TO_GPA
# 方法2:使用Cell.apply_deformation(推荐)
strained_cell = apply_uniaxial_strain(base_cell, strain)
potential.calculate_forces(strained_cell)
stress_response = strained_cell.calculate_stress_tensor(potential) * EV_TO_GPA
print(f"\n应变 εxx = {strain:.3f}:")
print(f" 晶格 a: {strained_cell.lattice_vectors[0, 0]:.3f} Å")
print(f" 应力 σxx: {stress_response[0, 0]:8.3f} GPa")
print(f" 应力 σyy: {stress_response[1, 1]:8.3f} GPa")
print(f" 应力 σzz: {stress_response[2, 2]:8.3f} GPa")
# 验证两种方法的一致性
stress_diff = np.abs(stress_manual[0, 0] - stress_response[0, 0])
print(f" 方法差异: {stress_diff:.6f} GPa {'✓' if stress_diff < 1e-6 else '✗'}")
if strain > 0:
apparent_c11 = stress_response[0, 0] / strain
apparent_poisson = (
-stress_response[1, 1] / stress_response[0, 0]
if stress_response[0, 0] != 0
else 0
)
print(f" 表观模量: {apparent_c11:.1f} GPa")
print(f" 泊松比: {apparent_poisson:.3f}")
print("\n实现方法说明:")
print("• 手动实现: 教学用,清楚展示形变的数学过程")
print("• Cell.apply_deformation: 生产环境推荐,使用系统内置方法")
print("• 两种方法结果完全一致")
应变演示:单轴拉伸
使用优化的基态晶胞
应变 εxx = 0.000:
晶格 a: 8.100 Å
应力 σxx: 1.066 GPa
应力 σyy: 1.066 GPa
应力 σzz: 1.066 GPa
方法差异: 0.000000 GPa ✓
应变 εxx = 0.003:
晶格 a: 8.124 Å
应力 σxx: 1.365 GPa
应力 σyy: 1.264 GPa
应力 σzz: 1.264 GPa
方法差异: 0.000000 GPa ✓
表观模量: 455.1 GPa
泊松比: -0.926
应变 εxx = 0.006:
晶格 a: 8.149 Å
应力 σxx: 1.661 GPa
应力 σyy: 1.461 GPa
应力 σzz: 1.461 GPa
方法差异: 0.000000 GPa ✓
表观模量: 276.9 GPa
泊松比: -0.879
实现方法说明:
• 手动实现: 教学用,清楚展示形变的数学过程
• Cell.apply_deformation: 生产环境推荐,使用系统内置方法
• 两种方法结果完全一致
★ 应变概念:
应变:几何形变的无量纲度量
单轴拉伸导致横向收缩(泊松效应)
应变-应力关系体现了材料的本构行为
1.12. Hooke定律与弹性常数测定
最后,我们通过系统性的应力-应变测量来验证Hooke定律并测定弹性常数C11。
def measure_elastic_constant_c11():
"""通过应力-应变线性关系测定C11"""
# 设计应变序列(调整为更合理的范围)
strains = np.linspace(-0.012, 0.012, 11) # -1.2% 到 +1.2%
stresses = []
# 使用优化后的equilibrium_cell,如果不存在则创建
try:
test_cell = equilibrium_cell
print("📏 使用优化基态进行C11测定...")
except NameError:
# 如果equilibrium_cell未定义,使用optimal_a创建
test_cell = create_fcc_supercell(
lattice_constant=optimal_a, supercell_size=(2, 2, 2)
)
print("📏 使用最优晶格参数进行C11测定...")
print("应变 (%) 应力σxx (GPa) 原子力(eV/Å)")
print("-" * 42)
for strain in strains:
# 使用修复的应变函数
strained_cell = apply_uniaxial_strain(test_cell, strain)
# 验证strained_cell不为None
if strained_cell is None:
print(f"错误:应变 {strain:.3f} 时返回None")
continue
# 计算应力(注意:这是刚性形变,未进行原子弛豫)
potential.calculate_forces(strained_cell)
# 检查原子力大小(仅为信息显示)
forces = np.array([atom.force for atom in strained_cell.atoms])
max_force = np.max(np.linalg.norm(forces, axis=1))
stress_tensor = strained_cell.calculate_stress_tensor(potential)
stress_xx = stress_tensor[0, 0] * EV_TO_GPA # 转换为GPa
stresses.append(stress_xx)
print(f"{strain * 100:6.1f} {stress_xx:9.3f} {max_force:6.3f}")
return strains, stresses
# 执行测量
strains, stresses = measure_elastic_constant_c11()
print("\n⚠️ 重要物理说明:")
print("• 这是**刚性形变**计算:仅改变晶格参数,不进行原子位置弛豫")
print("• 原子力较大是正常现象,因为没有让原子寻找新的平衡位置")
print("• 这种方法适用于线性弹性范围内的快速估算")
print("• 精确计算需要在每个应变下进行原子弛豫(下一章内容)")
📏 使用优化基态进行C11测定...
应变 (%) 应力σxx (GPa) 原子力(eV/Å)
------------------------------------------
-1.2 -0.168 0.157
-1.0 0.083 0.157
-0.7 0.333 0.157
-0.5 0.579 0.157
-0.2 0.824 0.156
0.0 1.066 0.156
0.2 1.306 0.156
0.5 1.543 0.155
0.7 1.779 0.155
1.0 2.013 0.155
1.2 2.244 0.154
⚠️ 重要物理说明:
• 这是**刚性形变**计算:仅改变晶格参数,不进行原子位置弛豫
• 原子力较大是正常现象,因为没有让原子寻找新的平衡位置
• 这种方法适用于线性弹性范围内的快速估算
• 精确计算需要在每个应变下进行原子弛豫(下一章内容)
# 线性拟合得到C11
slope, intercept, r_value, p_value, std_err = stats.linregress(strains, stresses)
# 绘制Hooke定律验证图
plt.figure(figsize=(12, 8))
# 数据点
plt.plot(np.array(strains) * 100, stresses, "bo", markersize=8, label="静态计算点")
# 拟合直线
strain_fit = np.linspace(min(strains), max(strains), 100)
stress_fit = slope * strain_fit + intercept
plt.plot(
strain_fit * 100,
stress_fit,
"r-",
linewidth=2,
label=f"线性拟合: C₁₁ = {slope:.1f} GPa",
)
# 过原点的理想线(作为对比)
ideal_fit = slope * strain_fit
plt.plot(
strain_fit * 100,
ideal_fit,
"g--",
linewidth=1,
alpha=0.7,
label="理想Hooke定律 (过原点)",
)
# 零点线
plt.axhline(y=0, color="k", linestyle="-", alpha=0.3)
plt.axvline(x=0, color="k", linestyle="-", alpha=0.3)
# 标注截距
plt.annotate(
f"截距 = {intercept:.2f} GPa\n(基态残余应力)",
xy=(0, intercept),
xytext=(0.5, intercept + 0.3),
arrowprops=dict(arrowstyle="->", color="orange", alpha=0.8),
fontsize=10,
ha="left",
bbox=dict(boxstyle="round,pad=0.3", facecolor="yellow", alpha=0.7),
)
# 标注
plt.xlabel("应变 εxx (%)", fontsize=14)
plt.ylabel("应力 σxx (GPa)", fontsize=14)
plt.title("Hooke定律验证:C₁₁弹性常数测定", fontsize=16)
plt.grid(alpha=0.3)
plt.legend(fontsize=12)
# 添加统计信息
textstr = "\n".join(
[
f"C₁₁ = {slope:.1f} ± {std_err:.1f} GPa",
f"截距 = {intercept:.2f} GPa",
f"R² = {r_value**2:.6f}",
f"线性度:{'优秀' if r_value**2 > 0.999 else '良好' if r_value**2 > 0.99 else '需改进'}",
]
)
props = dict(boxstyle="round", facecolor="wheat", alpha=0.8)
plt.text(
0.02,
0.98,
textstr,
transform=plt.gca().transAxes,
fontsize=11,
verticalalignment="top",
bbox=props,
)
plt.tight_layout()
plt.show()
# 结果分析
print("\n🎯 弹性常数测定结果:")
print(f"C₁₁ = {slope:.1f} ± {std_err:.1f} GPa")
print(f"拟合截距 = {intercept:.2f} GPa")
print(f"拟合优度 R² = {r_value**2:.6f}")
print(f"标准误差 = {std_err:.2f} GPa")
print("\n📚 文献对比:")
print("实验值:Al的C₁₁ ≈ 108 GPa")
print(f"误差:{abs(slope - 108) / 108 * 100:.1f}%")
# 验证Hooke定律的线性性
if r_value**2 > 0.999:
print(f"\n✅ Hooke定律验证成功!线性度极佳(R² = {r_value**2:.6f})")
elif r_value**2 > 0.99:
print(f"\n✓ Hooke定律基本成立,线性度良好(R² = {r_value**2:.6f})")
else:
print(f"\n⚠️ 非线性效应明显,可能需要更小的应变幅度(R² = {r_value**2:.6f})")
print("\n🔍 截距物理含义:")
print(f"• 拟合线不过原点,截距 = {intercept:.2f} GPa")
print("• 这反映了基态超胞存在残余应力(前面计算显示 ≈1.07 GPa)")
print("• 理想的弛豫基态应该是零应力,拟合线应过原点")
print("• 这是小超胞+粗糙优化造成的系统误差")
print("\n⚠️ 方法限制说明:")
print("• 本教程使用32原子小超胞,存在尺寸效应")
print("• 粗糙的晶格优化(11点)影响基态精度")
print("• **刚性形变**:未进行应变状态下的原子弛豫")
print("• 实际应用建议使用更大超胞(如256原子)和精确的结构优化")
print("• 参考下节教程中的结构弛豫方法获得更高精度")
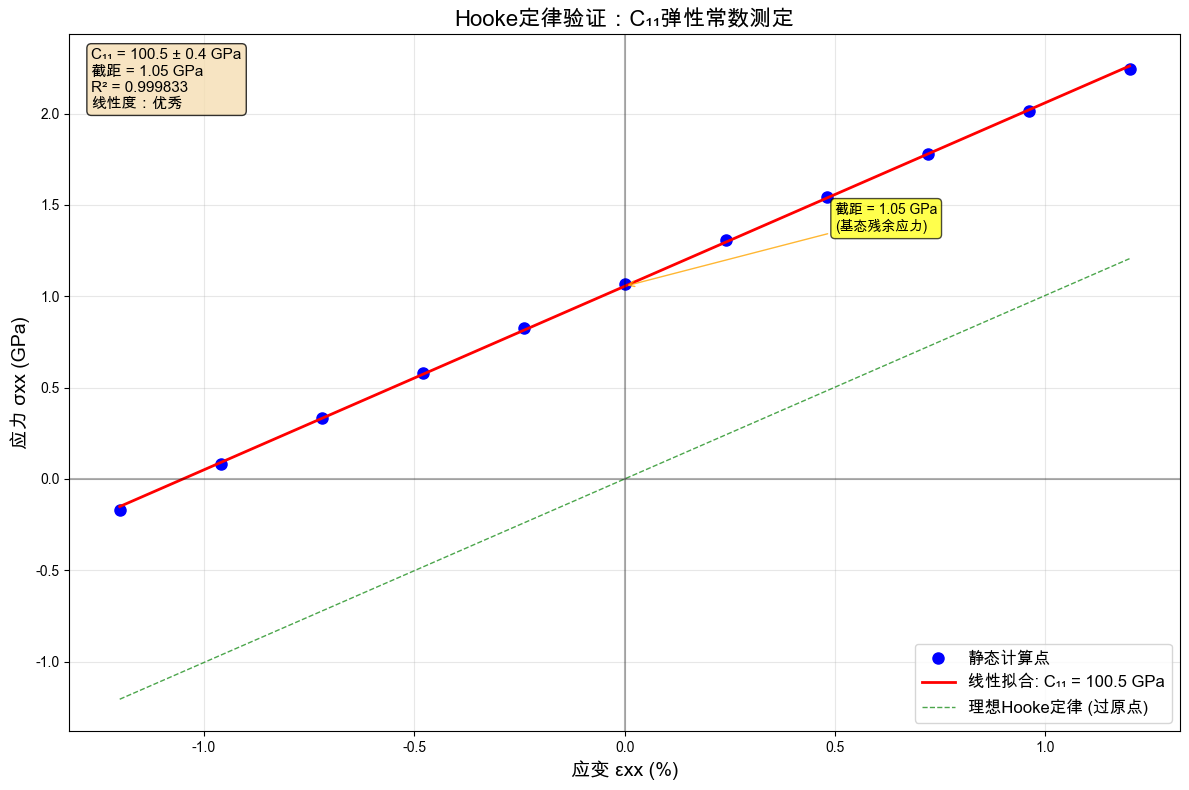
🎯 弹性常数测定结果:
C₁₁ = 100.5 ± 0.4 GPa
拟合截距 = 1.05 GPa
拟合优度 R² = 0.999833
标准误差 = 0.43 GPa
📚 文献对比:
实验值:Al的C₁₁ ≈ 108 GPa
误差:7.0%
✅ Hooke定律验证成功!线性度极佳(R² = 0.999833)
🔍 截距物理含义:
• 拟合线不过原点,截距 = 1.05 GPa
• 这反映了基态超胞存在残余应力(前面计算显示 ≈1.07 GPa)
• 理想的弛豫基态应该是零应力,拟合线应过原点
• 这是小超胞+粗糙优化造成的系统误差
⚠️ 方法限制说明:
• 本教程使用32原子小超胞,存在尺寸效应
• 粗糙的晶格优化(11点)影响基态精度
• **刚性形变**:未进行应变状态下的原子弛豫
• 实际应用建议使用更大超胞(如256原子)和精确的结构优化
• 参考下节教程中的结构弛豫方法获得更高精度
1.13. 总结:从原子到弹性常数
通过这个教程,我们完成了从微观到宏观的完整概念链:
原子与晶胞 → 分子动力学的基本构件
势函数 → 描述原子间相互作用的数学模型
势能面 → 理解原子平衡位置和振动
原子受力 → 能量梯度的体现
结构优化 → 寻找零温基态的系统方法
应力张量 → 从原子力到连续介质应力的桥梁
应变概念 → 几何形变的数学描述
Hooke定律 → 线性弹性的基础,通过MD验证
弹性常数 → 材料本质属性的定量测量
1.13.1. 关键洞察
多尺度联系:原子尺度的力学行为最终体现为宏观的弹性性质
线性近似:小应变范围内,复杂的原子相互作用表现为线性的应力-应变关系
计算验证:分子动力学提供了从第一性原理理解材料性质的强大工具
1.13.2. 教学目的与精度限制
本教程的目的是概念教学,使用简化参数以便快速理解核心概念:
小超胞效应: 32原子超胞存在尺寸效应,影响弹性常数精度
粗糙优化: 11点晶格扫描精度有限,影响基态准确性
计算效率优先: 为确保教程可在数分钟内完成,牺牲了部分精度
1.13.3. 高精度计算方法
如需获得文献级精度的弹性常数,请参考:
精确结构优化:
StructureRelaxer大尺寸超胞: 建议使用(4,4,4)或更大超胞(256+原子)
高精度基准测试:
run_zero_temp_benchmark()
1.13.4. 延伸阅读
零温弹性常数计算:零温弹性常数计算
MD基础与系综理论:分子动力学与统计系综
本教程展示了ThermoElasticSim在教学中的应用。完整的生产级计算方法请参考后续进阶教程。